The problem of computing native-like conformations when no average structure is available or descriptive of the protein native state is more challenging. Addressing this question, however, is becoming increasingly necessary due to the rampantly growing amount of genomic sequences and the lagging structural classification of sequence data. Extracting in silico the ensemble of functionally-relevant structures from protein sequences is a central challenge to computational molecular biology.
The Native state characterization of Cyclic Peptides (NcCYP) method was developed to address this problem for short (10-31 amino acids long) protein sequences with a characteristic geometric constraint: cyclization. The method limits the amount of a priori information to (i) amino acid sequence and (ii) a geometric constraint that results from cyclization in the native state of cyclic cysteine-rich peptides. The focus on cyclic cysteine-rich peptides is duly placed: these peptides are extremely robust, stable, and exhibit a rich array of diverse therapeutic properties.
The NcCYP exploits cyclization in cyclic peptides as a geometric constraint that lowers the dimensionality of the conformational space relevant for the native state. The search for native conformations proceeds in two stages: A broad view of cyclic low-energy conformations is first obtained. Second, conformations representative of emerging energy minima are iteratively used as references to compute more conformations and enrich the explored space with more energy minima until no lower-energy minima are obtained.
The search is conducted at multiple resolutions in order to extract all-atom detail native conformations efficiently. The broad view is obtained over a coarse-grained conformational space, where only the peptide backbone is explicitly modeled. The iterative enrichment of the conformational space and exploration of emerging low-energy minima is conducted in an all-atom conformational space. In addition, since these peptides are very rich in cysteines and disulfide bonds, a novel heuristic is proposed to compute an optimal cysteine arrangement into disulfide bonds in generated conformations.
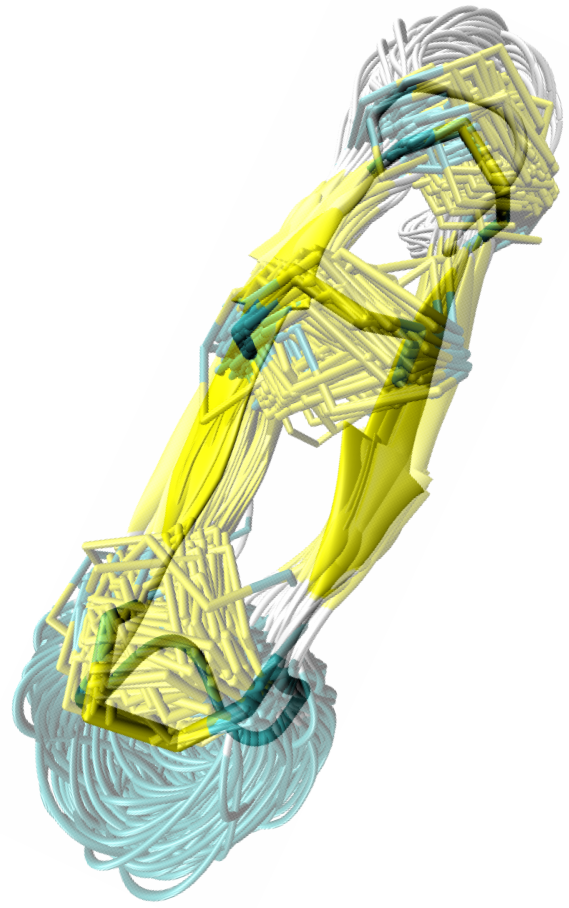
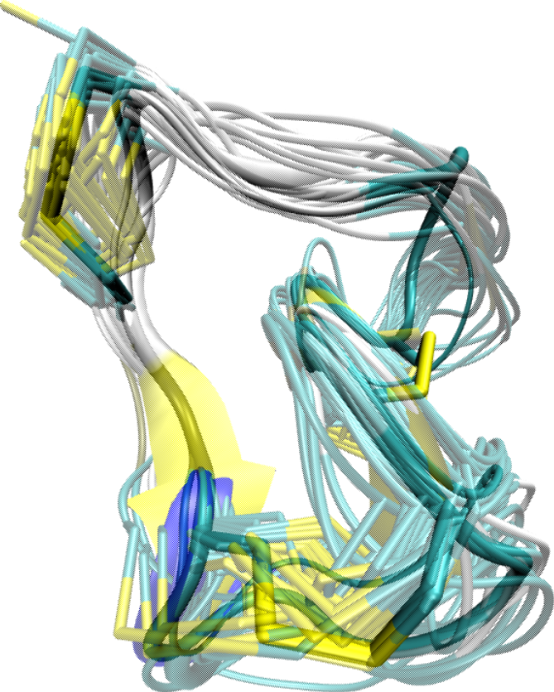
Applications of the NcCYP method to both naturally-occurring and engineered cyclic cysteine-rich peptides 20-30 amino acids long show that the method can obtain a comprehensive view of the conformational space relevant for the native state. The following figure shows a lower-dimensional embedding of the conformational space associated with low-energy conformations computed by the method from the RTD-1 sequence. The shown embedding is obtained through a non-linear dimensionality reduction technique known as SciMAP.


Left: In two-dimensional embedding color-coded with free-energy values, two local minima emerge (deep and light blue). Right: One-dimensional embedding shows a 10 RT unit separation in free energy values between the minima. The separation shows that only the deep blue minimum is significantly populated under native conditions.
Color-coding the embedding with energy values highlights conformational states associated with present minima. Two minima are obtained for RTD-1. The energy difference between the minima is significant enough to predict only one of them as being relevant under native conditions - the global minimum. The conformational ensemble associated with the local minimum is strikingly homogeneous, though conformations in NcCYP are sampled independently of one another. The conformational ensemble associated with this global minimum is strikingly similar to the NMR ensemble available for RTD-1. The method also obtains the correct disulfide-bond arrangement in the native state.
This work appears in: 1) Amarda Shehu, Lydia E. Kavraki, and Cecilia Clementi "Unfolding the Fold of Cyclic Cysteine-rich Peptides" Protein Science, 2008, 17(3):482-493.
On this Project:
-
Amarda Shehu
Lydia Kavraki
Cecilia Clementi
This project is completed.