

Characterizing functionally-relevant conformations in silico is particularly challenging when employing no structural or geometric information to localize the exploration. Yet, the problem of extracting from the amino-acid sequence native conformational subensembles of proteins with potentially multiple functional states is crucial to decoding the sequence-structure-function relationship in proteins.
The Multiscale Space Exploration (MuSE) method is recently proposed to efficiently explore the vast high-dimensional conformational space of a protein chain employing only knowledge of the protein's amino-acid sequence. MuSE is a multiscale method that proceeds in two stages. The method first obtains a broad view of the entire conformational space at a coarse-grained level of detail. In the second stage, the exploration focuses to few selected low-energy regions in the space.
In its first stage, the method searches a coarse-grained conformational space, employing structural databases to assemble low-resolution structures. The method adopts the fragment-based assembly of protein conformations, which is currently the most successful ab-initio approach in protein structure prediction. However, the proposed method focuses on computing not just one structure, but ensembles of native-like conformations that may be potentially diverse.
The fragment-based assembly is employed in the context of a simulated annealing exploration, which employs a coarse-grained force field to guide the assembly process. Most importantly, during the first stage of the exploration MuSE adds atomic detail on the fly to detect emerging energy minima possibly relevant in an all-atom view of the conformational space. This detail is stripped off to continue exploring the coarse-grained space. Atomistic refinement and further analysis of the explored conformational space is conducted in the second stage, after MuSE obtains a broad view of the coarse-grained conformational space relevant for the native state. Low-dimensional embedding highlights energy minima that are further populated by the method in all-atom detail.
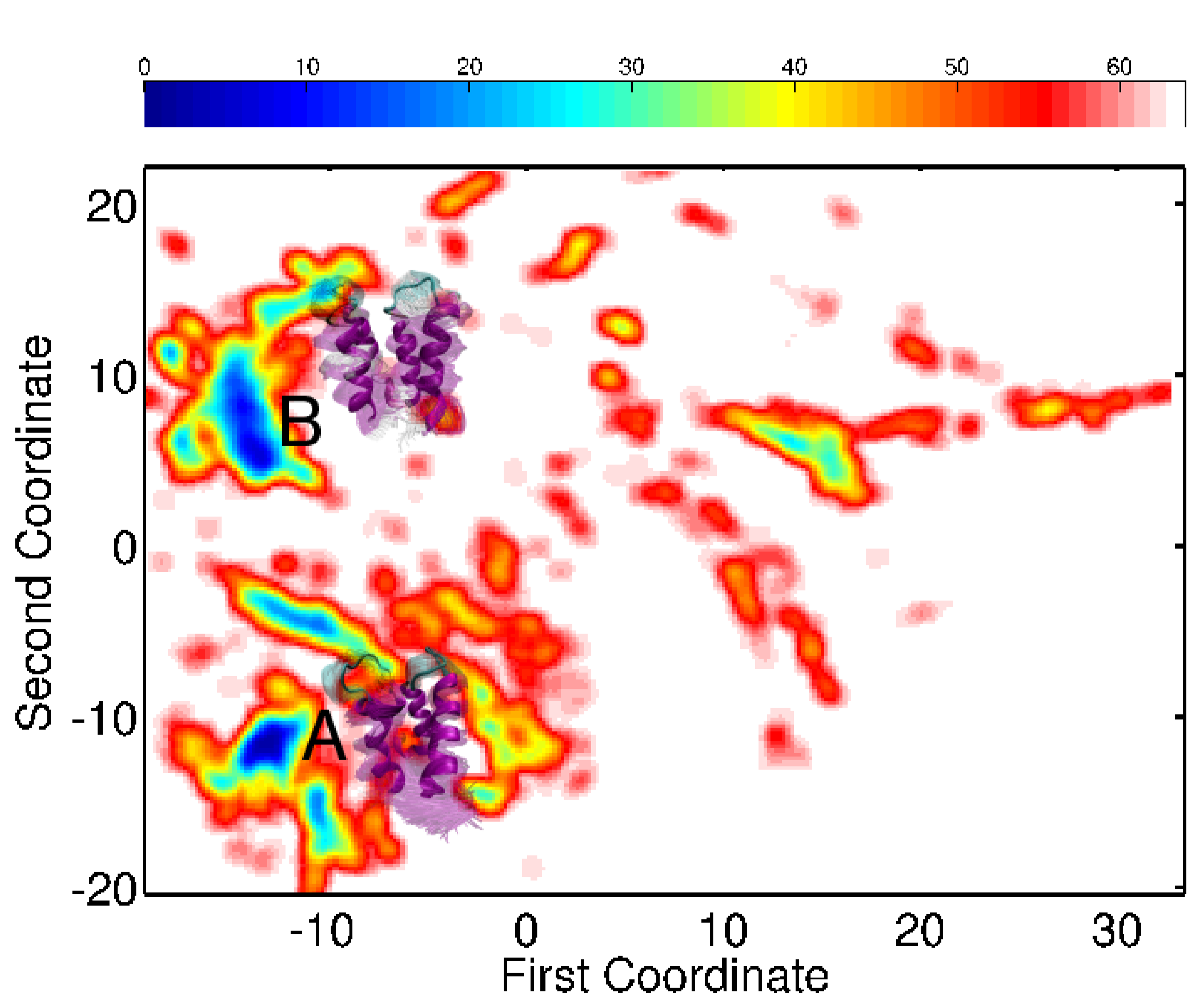
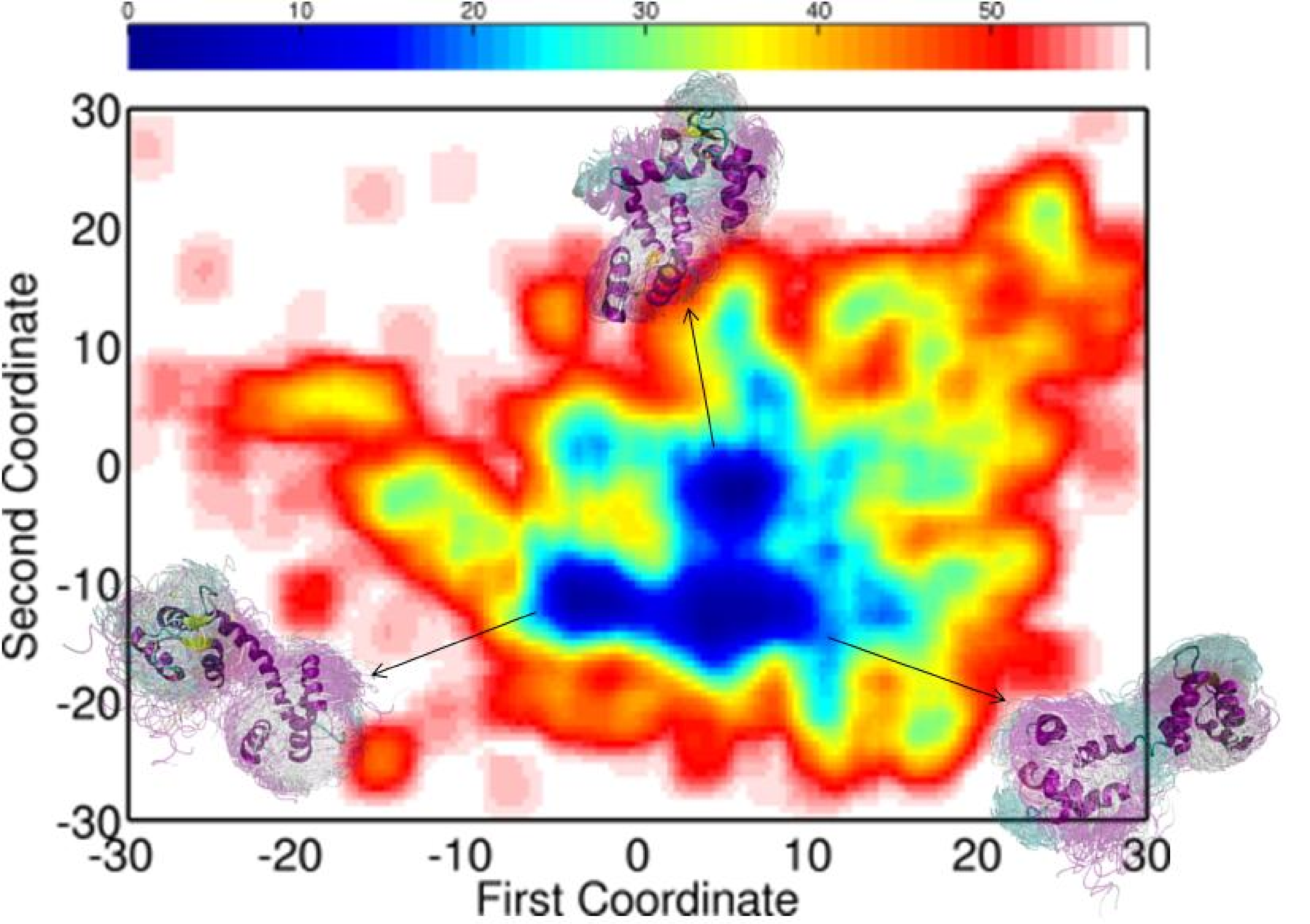
Embedding of the energy surfaces explored for the calbindin (left) and calmodulin (right) sequences reveal low-energy minima relevant for the native state. The conformational ensembles associated with the minima capture well the diverse functional states of each of the proteins.
Applications of the method on different protein sequences show that the lowest-energy all-atom conformational ensembles obtained capture well the diverse functional states populated by the proteins under under native conditions. These applications suggest that MuSE can predict functional motions for further testing and refinement in wet labs. Currently, adaptations of the method are being tested in the context of enhancing and improving protein structure prediction in CASP.
This work appears in: 1) Amarda Shehu, Lydia E. Kavraki, and Cecilia Clementi "Multiscale Characterization of Protein Conformational Ensembles" Proteins: Structure, Function, and Bioinformatics, 2009,76(4):837-851.
On this Project:
-
Amarda Shehu
Lydia Kavraki
Cecilia Clementi
This project is completed.