The high-dimensionality of the space associated with conformations of a protein chain poses a direct challenge on a search method; many parameters determine the positions of atoms in a protein conformation. Moreover, energetic interactions give rise to a multitude of local minima in the energy surface associated with the protein conformational space. While native conformations are associated with the lowest-energy basin(s) of the protein energy surface, how can a search algorithm efficiently locate these basins?
The Protein Ensemble Method (PEM) was developed to compute low-energy conformations around an experimentally-available (average) protein structure. The available structure can be employed as reference to denote the location of the global energy minimum and focus the search. Essentially, PEM divides the problem of computing structural fluctuations around the reference structure into independent (parallelizable) subproblems of computing fluctuations of consecutive overlapping fragments of the protein chain. Probabilistic exploration then samples conformations using analogies with geometrically-constrained kinematic chains.
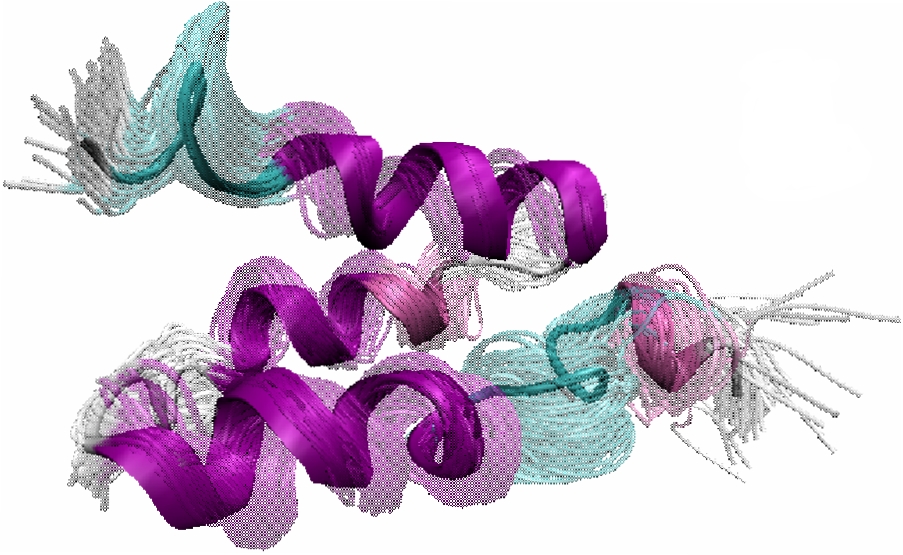
The protein chain is divided into consecutive fragments of significant overlap. This is illustrated below on the 123-aa chain of alpha-Lac. Sliding a fixed-length window of length 30 amino acids over the alpha-Lac chain defines 19 fragments where neighboring fragments overlap in 25 amino acids with one another. For each fragment, the FEM method is applied to obtain an ensemble of low-energy fragment conformations, which are pictorially illustrated by the ensembles inside each window. The final step of PEM combines fluctuations measured over the conformational ensembles of neighboring fragments to obtain a statistical picture of equilibrium fluctuations of the entire protein chain.
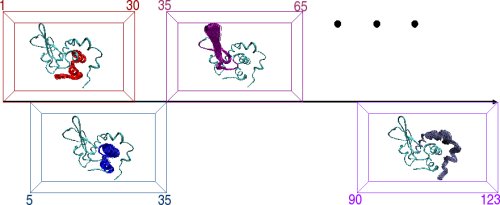
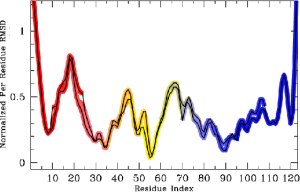
Left: An overview of PEM. Right: The end-result is illustrated through root-mean-squared-deviations (RMSD) measured over each amino acid of the chain. RMSD measurements obtained over different fragment ensembles are color-coded. Measurements of overlapping fragments are combined in a statistical mechanics framework to characterize the flexibility of the entire alpha-Lac chain.
PEM exploits locality, the fact that in proteins with non-concerted motions global information can be obtained by combining local information. This local-to-global strategy, known as a first-order approximation in biophysics, while limiting the domain of applications to proteins with non-concerted motions under native conditions, allows obtaining atomic fluctuations in silico. Applications of PEM to proteins of diverse lengths and native folds reproduce wet-lab data of broad (nanosecond-microseconds) time scales, as illustrated here for proteins like ubiquitin, protein G, and PAB.
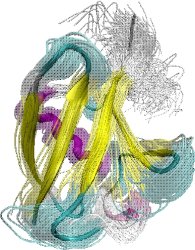
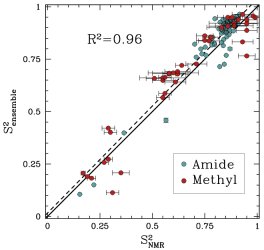
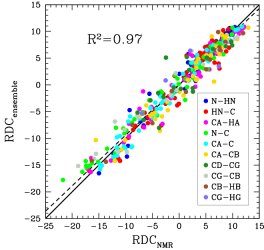
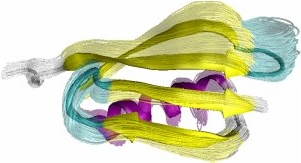
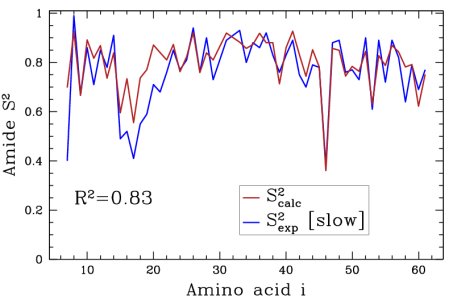
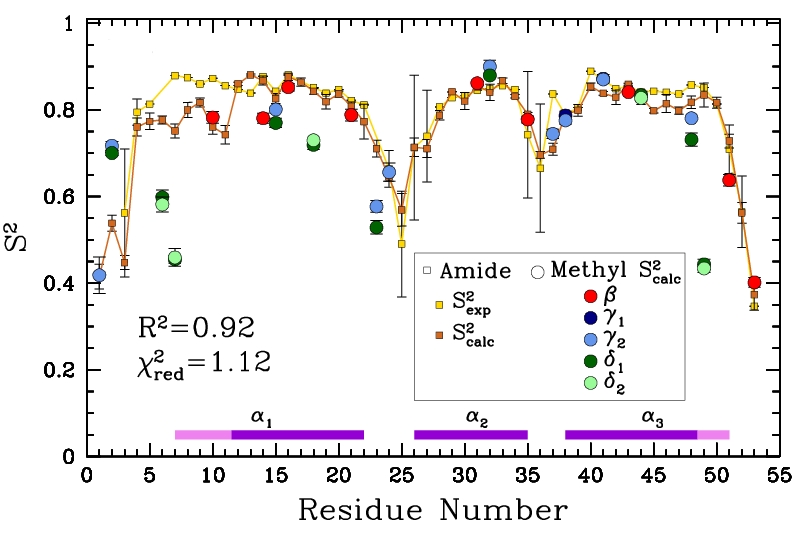
Obtained conformations for ubiquitin (in transparent) are superimposed over the native structure (in opaque). Amide and methyl order parameters (middle) and residual dipolar couplings (right) measured over PEM-obtained conformations agree very well with respective NMR data. This is significant, considering that nanoseconds-long MD simulations in explicit water reproduce NMR data poorly.
This work appears in: 1) Amarda Shehu, Cecilia Clementi, and Lydia E. Kavraki "Sampling Conformation Space to Model Equilibrium Fluctuations in Proteins" Algorithmica, 2007, 48(4):303-327; 2) Amarda Shehu, Lydia E. Kavraki, and Cecilia Clementi "On the Characterization of Protein Native State Ensembles" Biophysical Journal, 2007, 92(5):1503-1511; and 3) Amarda Shehu, Cecilia Clementi, and Lydia E. Kavraki "Modeling Protein Conformational Ensembles: From Missing Loops to Equilibrium Fluctuations" Proteins: Structure, Function, and Bioinformatics 2006, 65(1):164-179.
On this Project:
-
Amarda Shehu
Cecilia Clementi
Lydia Kavraki
This project is completed.